
Our long term goals are to understand the molecular mechanisms for the function and antigenic properties of proteins by analyzing the increasing amount of data from genomics, proteomics and metabolomics projects with novel bioinformatics tools.
Specific projects:
A) Structural Biology of Allergens:
Understanding the characteristics of allergenic proteins is important for guiding the treatment of patients, for reducing allergens in our food and air, and eventually for designing hypoallergenic proteins for use in foods and immunotherapy. My research group established a Structural Database of Allergenic Proteins (SDAP) (https://fermi.utmb.edu/SDAP/) that provides rapid, cross-referenced access to sequences, structures, and IgE epitopes of more than 900 allergens [1] [2] [3]. In a recent study we could demonstrate the high quality of our 3D models made available in SDAP [4]. For commercial information about SDAP, please click here to download the document .
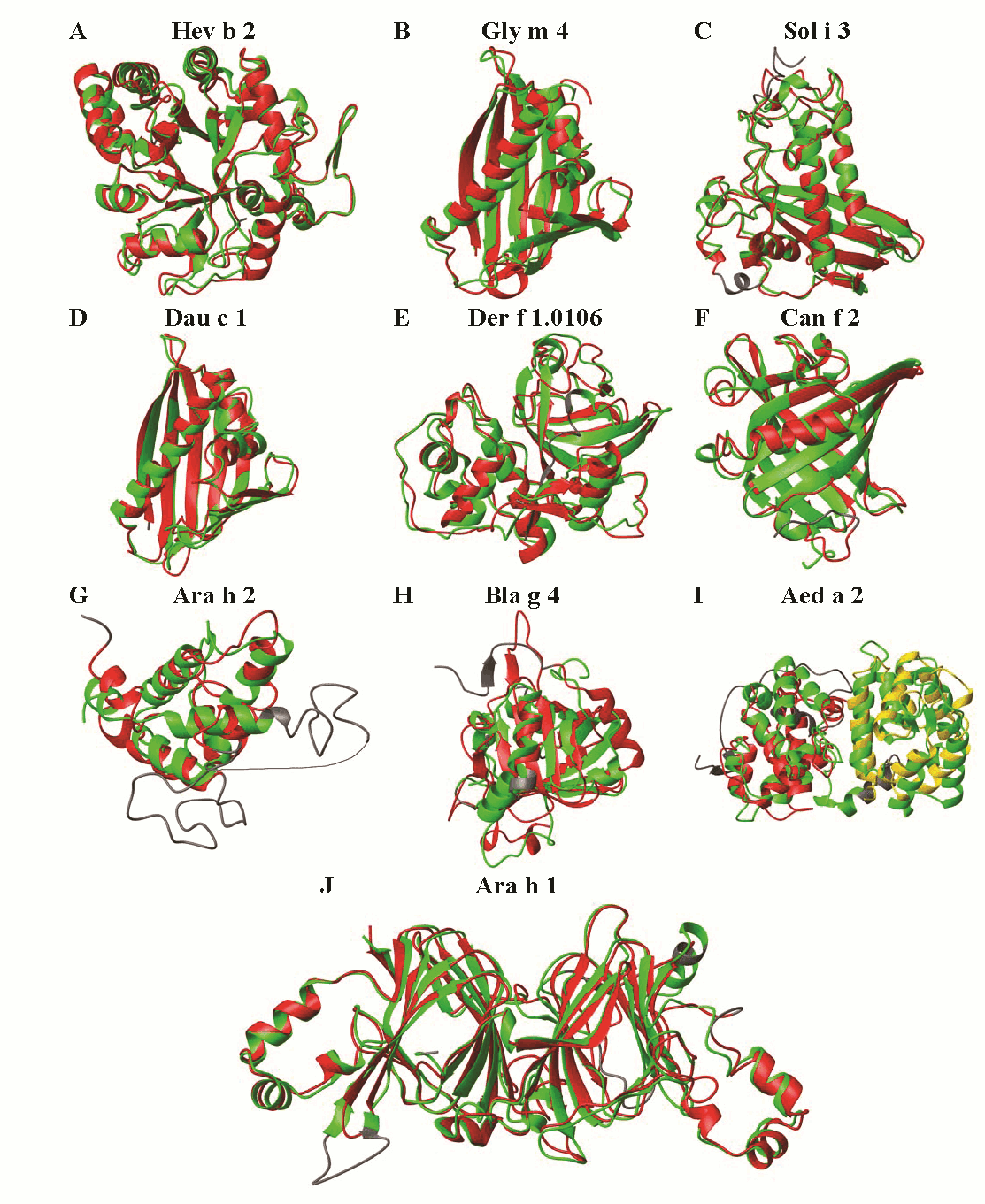
Figure : Model structures of allergeinc proteins in SDAP database. A sample of 3D models (red) which were predicted previously and later confirmed by experimental 3D structures (green) is shown in the figure on the right.
B) Structural Studies on Envelope Proteins of Alpha and Filo viruses:
Encephalitic alphaviruses, which include Eastern (EEEV), Western (WEEV) and Venezuelan Equine Encephalitis viruses (VEEV), cause significant disease in humans in all parts of the world and represent serious biological threats. In collaboration with colleagues at UTMB we focus on the detection and prediction of antigenic sites of the envelope proteins [5]. The results of this project will have impact on the design of new vaccines for alphaviruses with broad based protection. Several constructs of vaccine candidates are currently in the experimental test phase. A similar approach is currently in progress to give broad protection among the five distinct species of the Ebola virus [6], Zaire (EBOV), Sudan (SUDV), Tai Forest (TAFV), Reston (RESTV) and Bundibugyo (BDBV) virus.
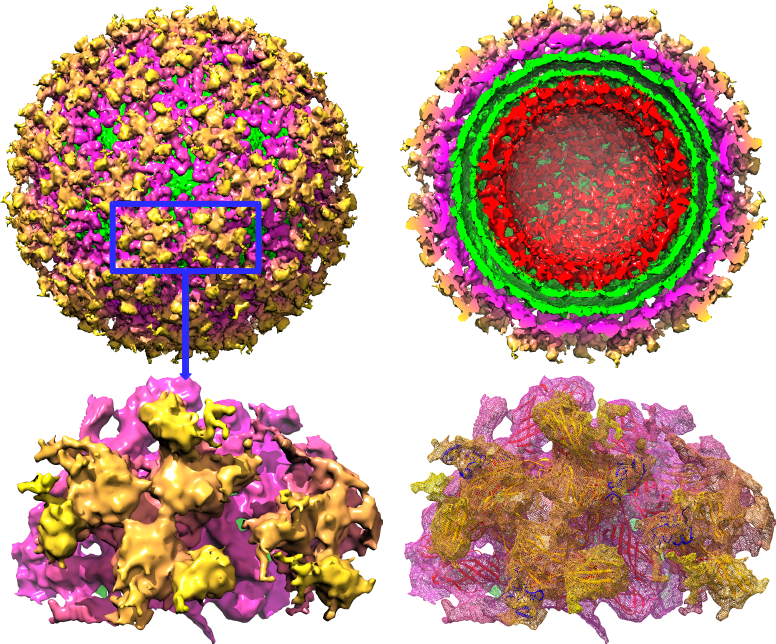
Figure : Structural organization of envelope protein E1 and E2 on WEEV surface
C) Development of Bioinformatics Tools for Conformational Epitopes Prediction:
Precise determination of conformational epitopes of neutralizing antibodies represents a key step in the rational design of novel vaccines. We developed a fully automated search method, EpiSearch [7] [8] that predicts the possible location of conformational epitopes from phage display experiments. New developments include prediction of conformational epitopes from 3D structures using InterProSurf [9] and differential sequence analysis by our PCPMer [10] [11] software suite. These software tools are applied to assess the potential allergenicity of proteins and designing novel vaccine candidates of emerging infectious diseases.
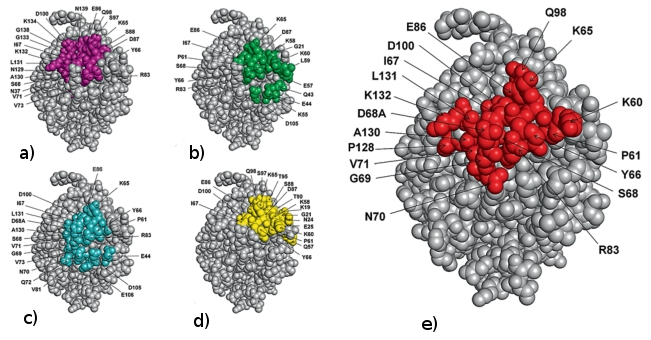
Figure : Comparison of predicted residues by EpiSearch (left) and experimentally observed antibody contact residues (right) in the X-ray co-crystal structure of the allergen Bla g 2.
D) Computational Methods for Potential Toxic Effects of Proteins:
Our goal is to provide novel, validated methods to rapidly assess the similarity of a novel protein to known protein toxins. This will allow biotechnology companies to evaluate the potential for adverse effect of novel engineered proteins early in the development cycle and mitigate the effect by specific amino acid substitutions. The quantitative analysis can also be used as supporting material for the endorsement of a final product to regulatory agencies.